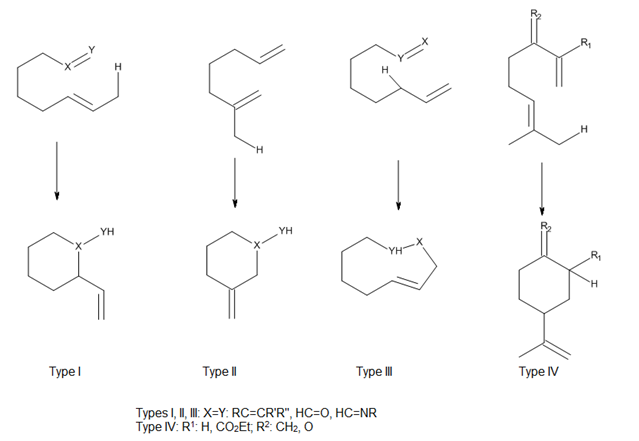
Intramolecular Diels-Alder and Alder Ene Reactions
Free download. Book file PDF easily for everyone and every device. You can download and read online Intramolecular Diels-Alder and Alder Ene Reactions file PDF Book only if you are registered here. And also you can download or read online all Book PDF file that related with Intramolecular Diels-Alder and Alder Ene Reactions book. Happy reading Intramolecular Diels-Alder and Alder Ene Reactions Bookeveryone. Download file Free Book PDF Intramolecular Diels-Alder and Alder Ene Reactions at Complete PDF Library. This Book have some digital formats such us :paperbook, ebook, kindle, epub, fb2 and another formats. Here is The CompletePDF Book Library. It's free to register here to get Book file PDF Intramolecular Diels-Alder and Alder Ene Reactions Pocket Guide.
Contents:
Diels—Alder reaction is one of the most fundamental synthetic transformations in organic chemistry for the construction of highly complex natural product scaffold with up to four contiguous stereocenters. Within this field, our research group have investigated the D-A reactions of 4-vinyl-imidazoles to target structures related to marine alkaloids.
The aim of which was to obtain bioactive molecules, based on the pyrrolo [3,4- a ]carbazole-1,3 2 H -dione framework Scheme 1.
Indolecarboxaldehyde was protected using p -toluenesulfonyl chloride followed by conversion to 1-tosylalkenyl-1 H -indoles 2a-b via a Wittig reaction. The Diels-Alder adducts 3a-3b were synthesised from the reaction of the corresponding 1-tosylalkenyl-1 H -indoles 2a-b with N -methylmaleimide 1. Scheme 2: Intermolecular D-A reaction of two different dienes.
We are currently investigating the optimisation of the Diels-Alder reactions of compound 2b and examining the subsequent ene chemistry. This reaction has been used for the synthesis of recifeiolide, ricinelaidic acid, and the insect pheromones E,E -8,dodecadienyl acetate and E -9,dodecadienyl acetate. Duncia, J. EtAlCl 2 catalyzes the ene reactions of a-substituted acrylate esters with trans 1,2-di- and trisubstituted alkenes.
The reactions are regio- and stereoselective. The ester group adds endo and a hydrogen is transferred selectively from the alkyl group syn to the vinylic hydrogen. Methyl a-chloroacrylate, a-bromoacrylate, acetamidoacrylate, and methacrylate, ethyl a-bromomethylacrylate, and dimethyl itaconate were explored. Dimethylaluminum chloride, which is a mild Lewis acid and a proton scavenger, catalyzes the ene reactions of aliphatic and aromatic aldehydes with alkenes containing a disubstituted vinylic carbon.
In the first, the isotoluene rearomatizes to provide polycyclic products like 32a - o Table 1 in a process explained by water-mediated proton shuttling. Corey, E. Having thus established a background point, this update plans to take the reader into the newer possibilities that have become fact in the last decade. You're using an out-of-date version of Internet Explorer. Ene reaction of arynes with alkynes. Hoffman HMR.
Proton-initiated rearrangements do not occur, since the alcohol-Lewis acid complex formed in the ene reaction reacts rapidly to give methane and a nonacidic aluminum alkoxide. This final point was reinforced by the outcome of our attempt to use the Lautens strategy cf. The biaryl 35 was formed, but in only ca. Products resulting from reduction 36 17 or amine trapping 37 of the intermediate aryne were formed predominantly. We have performed studies that bear on the mechanism of the final rearomatization event [cf.
Thermal, suprafacial, unimolecular reorganization of isotoluene to toluene is symmetry-forbidden, and the antarafacial variant of this 1,3-sigmatropic rearrangement is geometrically unfavorable 21 , if not untenable. Thus, the 1,3-hydrogen atom migration event typically requires catalysis We speculated that adventitious water might be acting as a proton shuttle to promote conversion of 31 to 32 in a fashion like that known for keto-enol tautomerization To test this idea, we heated triyne 38 in the presence of D 2 O specifically, D 2 O-saturated chloroform. The dominant product, 32e-d 1 Fig.
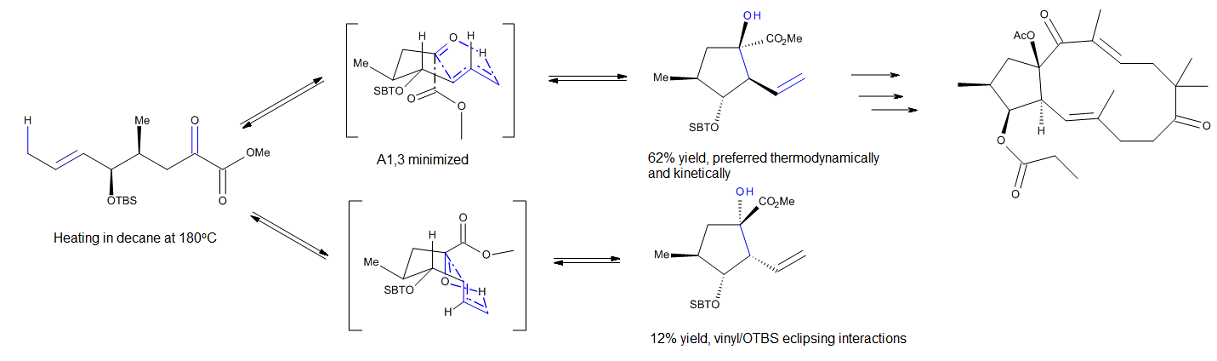
Intramolecular Diels-Alder – 1,3,9-decatrien-8-one
A complementary experiment was performed in which the trideuterated substrate d 3 was heated, this time in the presence of H 2 O. In this case, the dominant product was 32e-d 2 Fig. These results are best explained by a rearomatization process like that portrayed in the isotoluene intermediate We next reasoned that because the isotoluene intermediate 39 formed via the initial aromatic ene reaction was sufficiently long-lived to encounter water, it might be possible to trap this species in an even more productive manner by an added external enophile.
Indeed, when a 1,2-dichloroethane solution of the trideuterated substrate d 3 was heated in the presence of maleic anhydride 24 Fig. The successful transformation of 38 to 41 via 39 Fig. This sequence is enabled because the isotoluene intermediate 39 is still endowed with a considerable portion of the potential energy embodied by the three alkyne units in the initial HDDA substrate The scope of this three-stage cascade process can be seen from the examples shown in Table 2.
Although we do not know the exact origin of this selectivity, relative asymmetric induction in this case can occur at either or both of two stages. The aromatic ene substrate 44 can cyclize to either of the diastereotopic re and si faces at its two equivalent ortho -carbon atoms, and the approach of the Alder enophile to the isotoluene intermediate 43 can occur via either an endo or exo geometry. In conclusion, by capitalizing on the reagent-free, aryne-generating HDDA reaction and taking guidance from computational chemistry, we designed substrates that reveal the generality of a heretofore rare and elusive type of ene reaction in which an arene bearing a benzylic C—H bond functions as the ene donor.
Thus, two complementary overall transformations have emerged. In the first, the isotoluene rearomatizes to provide polycyclic products like 32a - o Table 1 in a process explained by water-mediated proton shuttling. In the second, the reactive isotoluene is further engaged by an external enophile to give products of yet greater structural complexity cf. This ene-upon-ene cascade reaction involves the overall formation of four carbon-carbon bonds and three rings, requires no external reagents, and generates no byproducts. The discovery of this efficient aromatic ene reaction further attests to the importance of a key feature of the HDDA cycloisomerization—namely, its ability to deliver aryne intermediates in the absence of the potentially interfering reagents that typically accompany aryne formation by classical methods.
Possibilities are under investigation here. Full experimental procedures for preparation of and complete spectroscopic characterization data for all new compounds aromatic ene substrates and products and a description of the computational protocols and results can be found in the Supplementary Information. A solution of the triyne precursor in the indicated solvent ca.
The product was purified by chromatography on silica gel after the indicated time. A solution of the triyne precursor in the indicated solvent 0. Unless otherwise noted, the product was purified by chromatography on silica gel after the indicated time. Author Contributions D. N and T.
Supplementary information and chemical compound information are available in the online version of the paper. Reprints and permissions information is available online at www. Competing financial interest The authors declare no competing financial interest. National Center for Biotechnology Information , U. Nat Chem. Author manuscript; available in PMC Dec Dawen Niu 1 and Thomas R.
Hoye 1.
Catalysis of Diels-Alder Reactions.
Thomas R. Author information Copyright and License information Disclaimer.
Correspondence and requests for materials should be addressed to: T. Copyright notice.
The publisher's final edited version of this article is available at Nat Chem. See other articles in PMC that cite the published article. Associated Data Supplementary Materials 1. Open in a separate window.
- Desert locust management: a time for change
- Beethovens Symphonies: An Artistic Vision
- The Mathematical Theory of Finite Element Methods
- Cultural Perspectives on the Mathematics Classroom
- Dogs of Courage: The Heroism and Heart of Working Dogs Around the World
- Revelation: Gods Gift of Hope (Six Weeks with the Bible)